Paul D. Ray, Bo-Wen Huang, and Yoshiaki Tsuji, Cell Signal, May 2012,: 24(5): 981-990.
Here is a direct link to the file: https://dx.doi.org/10.1016%2Fj.cellsig.2012.01.008
1. Introduction
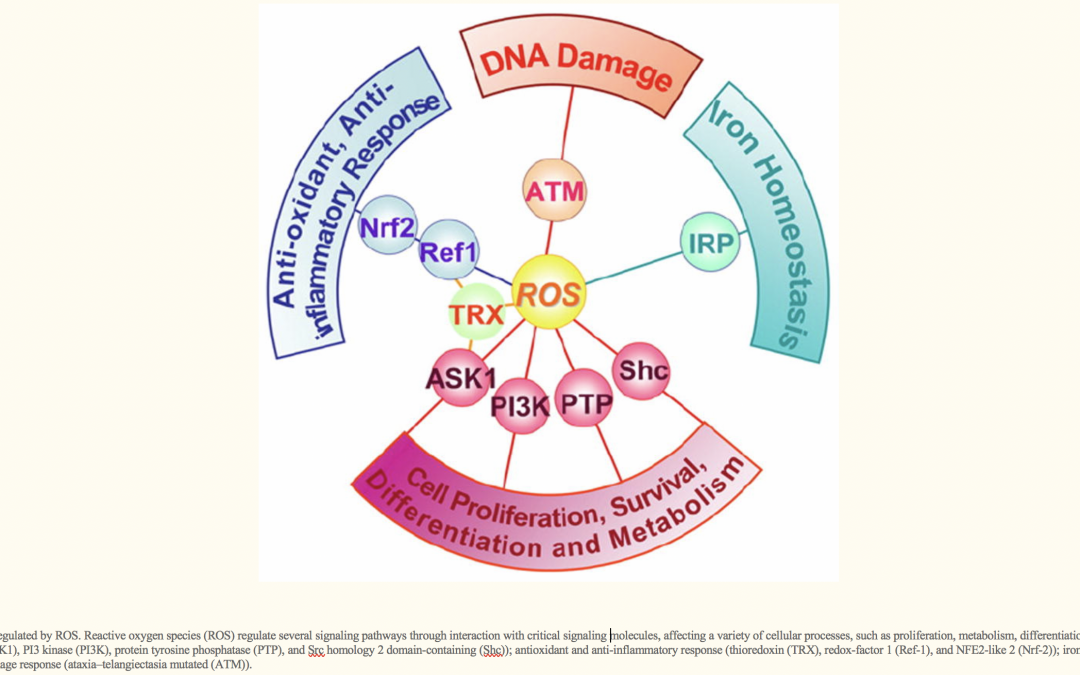
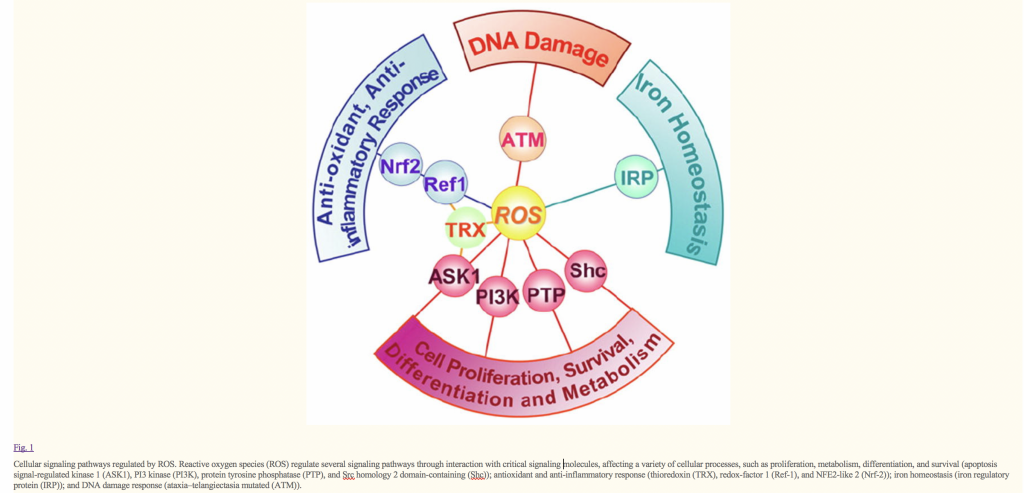
Reactive oxygen species (ROS), such as superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), consist of radical and non-radical oxygen species formed by the partial reduction of oxygen. Cellular ROS ( reactive oxygen species ), are generated endogenously as in the process of mitochondrial oxidative phosphorylation, or they may arise from interactions with exogenous sources such as xenobiotic compounds. When ROS overwhelm the cellular antioxidant defense system, whether through an increase in ROS levels or a decrease in the cellular antioxidant capacity, oxidative stress occurs. Oxidative stress results in direct or indirect ROS-mediated damage of nucleic acids, proteins, and lipids, and has been implicated in carcinogenesis [1], neurodegeneration [2,3], atherosclerosis, diabetes [4], and aging [5]. However, ROS involvement in the pathogenesis of disease states is not confined to macromolecular damage. There is increasing evidence that ROS signaling contributes to disease. For example, ROS have been shown to promote tumor metastasis through gene activation [6]. While there exists ample evidence demonstrating the role of ROS in regulating cellular signaling pathways, the question that is raised is exactly how do ROS initiate cellular signaling? The “oxidative interface” is that boundary between ROS and the signaling molecules they activate; that is, the figurative region that describes how ROS directly activate oxidative stress-responsive pathways. This review seeks to explore the oxidative interface between ROS and a functionally broad selection of cellular signaling pathways regulating a variety of cellular processes…